Quantitative Single-Cell Biology of the Immune System (Grün Lab)
Research
Mission
Cell fate decision is essential for the emergence of a complex multicellular organism starting from a single pluripotent zygote, and perpetually occurs during adult life to maintain organ tissues. The robustness of cell fate decision is remarkable given the stochastic interaction of hundreds of thousands of molecules in each cell. It has remained a central open question how signals from the microenvironment are integrated with stochastic processes in a cell to control cell fate decision in normal tissues and upon perturbations due to disease or tissue damage. Our lab investigates these questions by combining single-cell resolution experimental methods with computational methods involving machine learning and mathematical modeling.
Models
We are focusing on development, disease, and regeneration of organ tissues to understand the driving forces and control mechanisms of cell fate decision. We investigate a variety of systems which prominently show constant turnover, lineage plasticity, and response to perturbations in terms of cell differentiation dynamics. (1) We investigate the role of the bone marrow niche in regulating blood cell differentiation during development, leukemia, and regeneration after immune- or stem cell therapy. (2) As an organ system with high regenerative capacity and epithelial lineage plasticity, we focus on the liver to investigate the regulation of multicellular organ architecture during development, damage, and regeneration. (3) As a complimentary organ system with limited regenerative capacity in the adult mammal, we study the heart. We investigate the regulation of scar formation by multicellular crosstalk in the cardiac niche, and study determinants and roadblocks tissue regeneration by cardiomyocyte dedifferentiation.
Across all model systems, we combine experimental animal models with the analysis of human patient-derived material, and physiological in vitro models such as 3D organoids to pursue a translational approach.
Methods
With the recent emergence of powerful single-cell resolution experimental methods, including multimodal single-cell sequencing and single-molecule-resolution spatial transcriptomics (e.g., 10x Xenium, MERSCOPE), it has become possible to profile the composition of tissues at the level of individual cells across molecular layers. By quantifying the genome-wide transcriptional profiles and the state of the chromatin, cell states and types can be annotated and differentiation trajectories reflecting cell state transitions can be inferred. Spatial methods based on in situ sequencing or highly multiplexed microscopic imaging permit the quantification of the expression of hundreds of genes in tissue sections, enabling the reconstruction of spatial tissue architecture and the inference of molecular interactions between co-localized cells. Paired with computational machine learning techniques tailored to such multimodal, large datasets, these approaches facilitate the integrative analysis of stochastic gene expression variability, i.e., biological noise (intrinsic cell fate determinants), and signaling pathways as well as other determinants (e.g., competition for metabolites) mediating interactions of neighboring cells (extrinsic cell fate determinants).
A specific focus of the lab is the role of gene expression variability across single cells during differentiation (Grün et al., Nature Methods, 2014; Grün, Nature Methods, 2020; Rosales-Alvarez et al., Genome Biology, 2023). It has been shown that transcription is frequently not a continuous process but occurs in bursts. This induces substantial cell-to-cell variability of mRNA levels, and the role of this so-called biological noise during cellular differentiation is not well understood.
Using single-cell transcriptomics in conjunction with other multimodal single-cell sequencing assays we aim to elucidate how biological gene expression noise changes during differentiation and how it is mechanistically regulated. Since single-cell sequencing still suffers from substantial technical noise we also utilize microscopic imaging of individual mRNAs in single cells (single-molecule FISH) to investigate gene expression variability with high specificity and sensitivity.
 count per cell, and residual variability, which is assumed to be of biological origin (Rosales-Alvarez et al., Genome Biology, 2023).")
Our lab develops computational methods for the quantitative analysis of single-cell sequencing data, such as inference and modeling of differentiation trajectories (Grün et al., Nature, 2015; Grün# et al., Cell Stem Cell, 2016; Herman et al., Nature Methods, 2018; Wang et al., Genome Biology, 2024), the quantification of gene expression noise within cells (Grün, Nature Methods, 2020; Rosales-Alvarez et al., Genome Biology, 2023), and for the integration of spatial transcriptomics and single-cell RNA-sequencing data to study interactions of cells in tissues (Agrawal et al., Nature Communications, 2024).
.")
We are also exploring novel deep learning approaches such as transformer architectures, deep graph-neural networks, and deep reinforcement learning for improved modeling of multimodal single-cell sequencing and spatial transcriptomics data.
. We used Cell-DRL to predict a novel reprogramming path from human cardiac fibroblasts to cardiomyocytes. Cell-DRL enables reconstruction of patient-specific disease trajectories.")
The liver is the major metabolic organ of the body. Liver malignancies, such as fatty liver disease, liver cancer, or Hepatitis, are on the rise, in particular, in western societies as a consequence of an unhealthy lifestyle. To repair tissue damage, the liver has developed an extraordinary regenerative capacity. In the past, a large body of research has emerged, revealing distinct pathways of regeneration. For instance, liver resection and acute liver damage was found to initiate proliferation of hepatocytes, while upon chronic liver damage or suppression of hepatocyte proliferation parenchymal cells regenerate from the bile duct epithelium. However, our understanding of the molecular and cellular processes controlling cell states in the liver during health and disease is far from complete. We have a particular focus on the role of epithelial lineage plasticity and progenitor populations (Aizarani et al., Nature, 2019).
With the recent availability of single-cell resolution spatial transcriptomics, it becomes possible to elucidate multicellular crosstalk underlying liver development, damage and regeneration. We are studying cell state dynamics at spatial resolution with a particular focus on the role of cell-cell communication across the epithelial, immune, and mesenchymal compartments involved in the control of cellular differentiation and cell state plasticity in the liver.
 undergo dynamic turnover and induce an Il17-response of gamma-delta-T cells, highlighting a critical function of rare unconventional lymphocytes in fibrosis (Thoman et al., Nature Communications, 2026).")
As a model of multi-lineage differentiation we investigate the hematopoietic system in the bone marrow.
We are studying differentiation of murine hematopoietic cell populations in the bone marrow, as well as differentiation of tissue-resident cells of the innate immune system (Herman et al., Nature Methods, 2018; Sagar et al., The EMBO Journal, 2020; Zeis et al., Immunity, 2020; Rosales-Alvarez et al., Genome Biology, 2023). Our aim is to elucidate the dependence of hematopoietic cell fate decision on the local microenvironment during development, disease, and regeneration. To obtain information on the interactions of hematopoietic cells with neighboring cells in the local microenvironment, we are integrating single-cell sequencing methods with imaging-based spatial transcriptomics.
To facilitate translation of our research into biomedically relevant therapeutic approaches, we are combining the analysis of mouse models and human patient material. In particular, we investigate bone marrow response to CAR T cell therapy and subsequent regeneration to acquire a better understanding of frequently occurring severe side effects. The goal of this study is the identification of novel molecular targets for improving the outcome of CAR T cell therapy by reducing dangerous side effects.
. These data were compared between patients with full recovery of hematopoiesis and patients with prolonged cytopenia, leading to the discovery of inflammatory signaling interactions of mesenchymal stromal cells, myeloid cells, and hematopoietic stem cells underlying prolonged cytopenia (Cheung et al., unpublished).")
The mammalian heart is capable of complete regeneration during neonatal stages and this capacity is lost after birth. In the adult heart, cardiac damage, e.g., resulting from myocardial infarction, leads to replacement of cardiomyocytes by scar tissue, which can compromise the contractile function of the heart muscle. The niche determinants that control subsequent phases of healing, i.e., early inflammation, generation of scar tissue, and maturation of the scar, are only incompletely understood. Moreover, the molecular roadblocks of cardiomyocyte dedifferentiation required for full cardiac regeneration are only partially studied. A detailed understanding of cell-intrinsic as well as extrinsic niche determinants required for cardiomyocyte dedifferentiation and proliferation could permit the development of novel therapies enabling cardiac regeneration in the adult heart.
We have established a spatio-temporal atlas of scar formation upon ischemic damage in the murine heart by integrating single-cell RNA-seq and spatial transcriptomics timecourse data (Chan et al., Nature Cardiovascular Research, 2025). We discovered essential niche interactions involving fibroblasts and macrophages required for the tight control of scar formation, and identified niche determinants of cardiomyocyte dedifferentiation. Based on our discoveries we are aiming to study specific molecular pathways controlling cardiomyocyte dedifferentiation to enable cardiac regeneration.
. These data led to the discovery of niche determinants of cardiomyocyte dedifferentiation, which could be exploited to develop new therapies for improving cardiac regeneration. Furthermore, dynamic crosstalk between fibroblasts and macrophages for mutual control of proliferation dynamics was discovered, which is critical for tight regulation of scar formation.")
Grün link pubmed: https://pubmed.ncbi.nlm.nih.gov
Selected Articles
LIST OF PUBLICATIONS
(*co-first authors, #co-corresponding authors, ## co-senior, **Lead author)
-
Thomann S, Hemmer H, Agrawal A, Basu S, Schaf J, Sagar, Imdahl F, Poth T, Tóth M, Zielinski CE, Poch T, Krause J, Rosenwald A, Breitkopf-Heinlein K, Rahbari N, Grün D (2026) An immunobiliary single-cell atlas resolves crosstalk between type 2 conventional dendritic cells and γδ T cells in cholangitis. Nature Communications 17: 3455
-
Taylor SA, Bader GD, MacParland S, Mullen AC, Andrews T, Cuenca AG, DasGupta R, Gehring AJ, Grün D, Guilliams M, Gulamhusein A, Henderson NC, Hirschfield G, Huppert SS, Itzkovitz S, Jiang ZG, Lauer GM, McGilvray I, Mysore KR, Pirola CJ, Quon G, Rahbari M, Regev A, Ricciuto A, Scott CL, Sharma A, Sookoian S, Tana MM, Teichmann SA, Vallier L, Vlachos IS, Wang B, Zhen M (2026) Towards a reference cell atlas of liver diversity over the human lifespan. Nature Reviews Gastroenterology & Hepatology 23(1): 97-109
-
Shing-Fung Chan A, Greiner J, Marschhäuser L, Brennan T A, Perez-Feliz S, Agrawal A, Hemmer H, Sinning K, Wing Lam Cheung J, Iqbal Z, Klesen A, Vico T A, Aprea J, Hilgendorf I, Seidel T, Vaeth M, Rog-Zielinska E A, Kohl P, Schneider-Warme F, Grün DSpatiotemporal dynamics of the cardioimmune niche during lesion repair Nature Cardiovascular Research 4(11): 1550–1572.
-
NiCo Identifies Extrinsic Drivers of Cell State Modulation by Niche Covariation Analysis Nature Communications15(1):10628.
-
Wang W, (2024) Scalable identification of lineage-specific gene regulatory networks from metacells with NetID. Genome Biology 25(1):275
-
Germain R, Van Allen EM, Trynka G, Tsang JS, Grün D, Kiemen AL, Shi Y, Pugh Bernard A, Nolan GP (2024) AI and Immunology Immunity 57(6):1177-1181.
- Simon-Chica A, Klesen A, Emig R, Chan A, Greiner J, Grün D, Lother A, Hilgendorf I, Rog-Zielinska EA, Ravens U, Kohl P, Schneider-Warme F, Peyronnet R (2024) Piezo1 stretch-activated channel activity differs between murine bone marrow-derived and cardiac tissue-resident macrophages Journal of Physiology 602(18):4437-4456.
-
Czech M, Schneider S, Peltokangas N, El Khawanky N, et al., Gerner RR##, Grün D##, Zeiser R## (2024) Lipocalin-2 expression identifies an intestinal regulatory neutrophil population during acute graft-versus-host disease Science Translational Medicine 16(735):eadi1501.
-
Ugur M, Labios RJ, Fenton C, Knöpper K, Jobin K, Imdahl F, Golda G, Hoh K, Grafen A, Kaisho T, Saliba AE, Grün D, Gasteiger G, Bajénoff M, Kastenmüller W (2023) Lymph node medulla regulates the spatiotemporal unfolding of resident dendritic cell networks Immunity 56(8):1778-1793.e10.
-
Appiah B, Fullio CL, Ossola C, Bertani I, Restelli E, Cheffer A, Polenghi M, Haffner C, Garcia-Miralles M, Zeis P, Treppner M, Bovio P, Schlichtholz L, Mas-Sanchez A, Zografidou L, Winter J, Binder H, Grün D, Kalebic N, Taverna E, Vogel T (2023) DOT1L activity affects neural stem cell division mode and reduces differentiation and ASNS expression. EMBO Reports e56233. doi: 10.15252/embr.202256233
- Rosales-Alvarez RE, Rettkowski J, Herman JS, Dumbović G, Cabezas-Wallscheid N, Grün D (2023) VarID2 quantifies gene expression noise dynamics and unveils functional heterogeneity of ageing hematopoietic stem cells. Genome Biology 24(1):1
- Kaltenbach L, Martzloff P, Bambach SK, Aizarani N, Mihlan M, Gavrilov A, Glaser KM, Stecher M, Thünauer R, Thiriot A, Heger K, Kierdorf K, Wienert S, von Andrian UH, Schmidt-Supprian M, Nerlov C, Klauschen F, Roers A, Bajénoff M, Grün D, Lämmermann T (2023) Slow integrin-dependent migration organizes networks of tissue-resident mast cells. Nature Immunology 24(6):915-924
- Hess I, Sagar, O Meara C, Grün D, Schorpp M, Boehm T (2022) Stage-specific and cell type-specific requirements of ikzf1 during haematopoietic differentiation in zebrafish. Scientific Reports 12(1):2140
- Kabat AM, Hackl A, Sanin DE, Zeis P, Grzes KM, Baixauli F, Kyle R, Caputa G, Edwards-Hicks J, Villa M, Rana N, Curtis JD, Castoldi A, Cupovic J, Dreesen L, Sibilia M, Pospisilik JA, Urban JF Jr, Grün D, Pearce EL, Pearce EJ (2022) Resident TH2 cells orchestrate adipose tissue remodeling at a site adjacent to infection. Science Immunology 7(76):eadd3263
- Cheng P, Zhao X, Katsnelson L, Camacho-Hernandez EM, Mermerian A, Mays JC, Lippman SM, Rosales-Alvarez RE, Moya R, Shwetar J, Grün D, Fenyo D, Davoli T (2022) Proteogenomic analysis of cancer aneuploidy and normal tissues reveals divergent modes of gene regulation across cellular pathways. Elife 11:e75227
- Zhang YW, Mess J, Aizarani N, Mishra P, Johnson C, Romero-Mulero MC, Rettkowski J, Schönberger K, Obier N, Jäcklein K, Woessner NM, Lalioti ME, Velasco-Hernandez T, Sikora K, Wäsch R, Lehnertz B, Sauvageau G, Manke T, Menendez P, Walter SG, Minguet S, Laurenti E, Günther S, Grün D, Cabezas-Wallscheid N (2022) Hyaluronic acid-GPRC5C signalling promotes dormancy in haematopoietic stem cells. Nature Cell Biology 24(7):1038-1048
- Nusser A, Sagar, Swann JB, Krauth B, Diekhoff D, Calderon L, Happe C, Grün D#, Boehm T# (2022) Developmental dynamics of two bipotent thymic epithelial progenitor types. Nature 606(7912):165-171
- Hochrein SM, Wu H, Eckstein M, Arrigoni L, Herman JS, Schumacher F, Gerecke C, Rosenfeldt M, Grün D, Kleuser B, Gasteiger G, Kastenmüller W, Ghesquière B, Van den Bossche J, Abel ED, Vaeth M (2022) The glucose transporter GLUT3 controls T helper 17 cell responses through glycolytic-epigenetic reprogramming. Cell Metabolism 34(4):516-532
- Friedrich C, Taggenbrock RLRE, Doucet-Ladevèze R, Golda G, Moenius R, Arampatzi P, Kragten NAM, Kreymborg K, Gomez de Agüero M, Kastenmüller W, Saliba AE, Grün D, van Gisbergen KPJM, Gasteiger G. (2021) Effector differentiation downstream of lineage commitment in ILC1s is driven by Hobit across tissues. Nature Immunology 22(10):1256-1267
- Clapes T, Polyzou A, Prater P, Sagar, Morales-Hernández A, Ferrarini MG, Kehrer N, Lefkopoulos S, Bergo V, Hummel B, Obier N, Maticzka D, Bridgeman A, Herman JS, Ilik I, Klaeylé L, Rehwinkel J, McKinney-Freeman S, Backofen R, Akhtar A, Cabezas-Wallscheid N, Sawarkar R, Rebollo R, Grün D, Trompouki E. (2021) Chemotherapy-induced transposable elements activate MDA5 to enhance haematopoietic regeneration. Nature Cell Biology 23(7):704-717
- Hensel N, Gu Z, Sagar, Wieland D, Jechow K, Kemming J, Llewellyn-Lacey S, Gostick E, Sogukpinar O, Emmerich F, Price DA, Bengsch B, Boettler T, Neumann-Haefelin C, Eils R, Conrad C, Bartenschlager R, Grün D, Ishaque N, Thimme R, Hofmann M. (2021) Memory-like HCV-specific CD8 + T cells retain a molecular scar after cure of chronic HCV infection. Nature Immunology 22(2):229-239
- Olah M, Menon V, Habib N, Taga MF, Ma Y, Yung CJ, Cimpean M, Khairallah A, Coronas-Samano G, Sankowski R, Grün D, Kroshilina AA, Dionne D, Sarkis RA, Cosgrove GR, Helgager J, Golden JA, Pennell PB, Prinz M, Vonsattel JPG, Teich AF, Schneider JA, Bennett DA, Regev A, Elyaman W, Bradshaw EM, De Jager PL. (2020) Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nature Communications 11(1):6129
- Probst S, Sagar, Tosic J, Schwan C, Grün D, Arnold SJ. (2020) Spatiotemporal sequence of mesoderm and endoderm lineage segregation during mouse gastrulation. Development 148(1):dev193789
- Ramamoorthy S, Kometani K, Herman JS, Bayer M, Boller S, Edwards-Hicks J, Ramachandran H, Li R, Klein-Geltink R, Pearce EL, Grün D, Grosschedl R. (2020) EBF1 and Pax5 safeguard leukemic transformation by limiting IL-7 signaling, Myc expression, and folate metabolism. Genes & Development 34(21-22):1503-19
- Zeis P, Lian M, Fan X, Herman JS, Hernandez DC, Gentek R, Elias S, Symowski C, Knöpper K, Peltokangas N, Friedrich C, Doucet-Ladeveze R, Kabat AM, Locksley RM, Voehringe D, Bajenoff M, Rudensky AY, Romagnani C, Grün#,** D, Gasteiger G#. (2020) In situ maturation and tissue adaptation of type 2 innate lymphoid cell progenitors. Immunity 53(4):775-792.e9
- Pessoa Rodrigues C, Herman JS, Herquel B, Valsecchi CIK, Stehle T, Grün D, Akhtar A. (2020) Temporal expression of MOF acetyltransferase primes transcription factor networks for erythroid fate. Science Advances 6(21): eaaz4815
- Sagar, Pokrovskii M, Herman JS, Naik S, Sock E, Lausch U, Wegner M, Tanriver Y, Littman DR, Grün D. (2020) Deciphering the Regulatory Landscape of γδ T Cell Development by Single-Cell RNA-Sequencing. EMBO Journal 39(13): e104159
- Sheikh BN, Guhathakurta S, Tsang TH, Schwabenland M, Renschler G, Herquel B, Bhardwaj V, Holz H, Stehle T, Bondareva O, Aizarani N, Mossad O, Kretz O, Reichardt W, Chatterjee A, Braun LJ, Thevenon J, Sartelet H, Blank T, Grün D, von Elverfeldt D, Huber TB, Vestweber D, Avilov S, Prinz M, Buescher JM, Akhtar A. (2020) Neural metabolic imbalance induced by MOF dysfunction triggers pericyte activation and breakdown of vasculature. Nature Cell Biology 22(7): 828-841
- Mereu E, Lafzi A, Moutinho C, Ziegenhain C, McCarthy DJ, Álvarez-Varela A, Batlle E, Grün D, Lau JK, Boutet SC, Sanada C, Ooi A, Jones RC, Kaihara K, Brampton C, Talaga Y, Sasagawa Y, Tanaka K, Hayashi T, Braeuning C, Fischer C, Sauer S, Trefzer T, Conrad C, Adiconis X, Nguyen LT, Regev A, Levin JZ, Parekh S, Janjic A, Wange LE, Bagnoli JW, Enard W, Gut M, Sandberg R, Nikaido I, Gut I, Stegle O, Heyn H. (2020) Benchmarking single-cell RNA-sequencing protocols for cell atlas projects. Nature Biotechnology 38(6): 747-755
- Sagar, Grün D. (2020) Deciphering cell fate decision by integrated single-cell sequencing analysis. Annual Review of Biomedical Data Science 3:1-22
- Derecka M, Herman JS, Cauchy P, Ramamoorthy S, Lupar E, Grün D, Grosschedl R. (2020) EBF1-deficient bone marrow stroma elicits persistent changes in HSC potential. Nature Immunology 21(3): 261-273
- Grün D. (2020) Revealing Dynamics of Gene Expression Variability in Cell State Space. Nature Methods 17(1): 45-49
- Sankowski R, Böttcher C, Masuda T, Geirsdottir L, Sagar, Sindram R, Seredenina T, Muhs A, Scheiwe C, Shah MJ, Heiland DH, Schnell O, Grün D#, Priller J#, Prinz M#. (2019) Mapping microglia diversity in the human brain through the integration of high-dimensional techniques. Nature Neuroscience 22(12): 2098-2110
- Honkoop H, de Bakker DE, Aharonov A, Kruse F, Shakked A, Nguyen PD, de Heus C, Garric L, Muraro MJ, Shoffner A, Tessadori F, Peterson JC, Noort W, Bertozzi A, Weidinger G, Posthuma G, Grün D, van der Laarse WJ, Klumperman J, Jaspers RT, Poss KD, van Oudenaarden A, Tzahor E, Bakkers J (2019) Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife 8: e50163
- Sheikh BN, Bondareva O, Guhathakurta S, Tsang TH, Sikora K, Aizarani N, Sagar, Holz H, Grün D, Hein L, Akhtar A (2019) Systematic Identification of Cell-Cell Communication Networks in the Developing Brain. iScience 21: 273-287
- Hummel JF, Zeis P, Ebert K, Fixemer J, Konrad P, Schachtrup C, Arnold SJ, Grün D, Tanriver Y (2019) Single-cell RNA-sequencing identifies the developmental trajectory of C-Myc-dependent NK1.1- T-bet+ intraepithelial lymphocyte precursors. Mucosal Immunology 13(2): 257-270
- Aizarani A, Saviano A, Sagar, Mailly L, Durand S, Herman JS, Pessaux P, Baumert TF#, Grün D# (2019) A Human Liver Cell Atlas reveals Heterogeneity and Epithelial Progenitors. Nature, 572(7768): 199-204
- Kolter J, Feuerstein R, Zeis P, Hagemeyer N, Paterson N, d'Errico P, Baasch S, Amann L, Masuda T, Lösslein A, Gharun K, Meyer-Luehmann M, Waskow C, Franzke CW, Grün D, Lämmermann T, Prinz M, Henneke P (2019) A Subset of Skin Macrophages Contributes to the Surveillance and Regeneration of Local Nerves. Immunity 50(6):1482-1497.e7
- Sagar, Grün D (2019) Lineage Inference and Stem Cell Identity Prediction Using Single-Cell RNA-Sequencing Data. Methods in Molecular Biology 1975:277-301
- Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, Scheiwe C, Nessler S, Kunz P, van Loo G, Coenen VA, Reinacher PC, Michel A, Sure U, Gold R, Grün D, Priller J, Stadelmann C, Prinz M. (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature 566 (7744): 388-392
- Costa Jordão MJ, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai YH, Tay TL, Schramm E, Armbruster S, Hagemeyer N, Groß O, Mai D, Çiçek Ö, Falk T, Kerschensteiner M, Grün D, Prinz M (2019) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 363 (6425). pii: eaat7554
- Tay TL, Sagar, Dautzenberg J, Grün D#, Prinz M# (2018) Unique microglia recovery population revealed by single-cell RNAseq following neurodegeneration. Acta Neuropathologica Communications 6(1): 87
- Costa F, Grün D#, Backofen R# (2018) GraphDDP: a graph-embedding approach to detect differentiation pathways in single-cell-data using prior class knowledge. Nature Communications 9(1): 3685
- Roovers EF, Kaaij LJT, Redl S, Bronkhorst AW, Wiebrands K, de Jesus Domingues AM, Huang HY, Han CT, Riemer S, Dosch R, Salvenmoser W, Grün D, Butter F, van Oudenaarden A, Ketting RF (2018) Tdrd6a Regulates the Aggregation of Buc into Functional Subcellular Compartments that Drive Germ Cell Specification. Developmental Cell 46(3): 285-301
- Grün D (2018) Revealing routes of cellular differentiation by single-cell RNA-seq. Current Opinion in Systems Biology 11: 9-17
- Boisset JC, Vivié J, Grün D, Muraro M, Lyubimova A, van Oudenaarden A (2018) Mapping the physical network of cellular interactions. Nature Methods 15(7): 547-553
- Lu TT, Heyne S, Dror E, Casas E, Leonhardt L, Boenke T, Yang CH, Sagar, Arrigoni L, Dalgaard K, Teperino R, Enders L, Selvaraj M, Ruf M, Raja SJ, Xie H, Boenisch U, Orkin SH, Lynn FC, Hoffman BG, Grün D, Vavouri T, Lempradl AM, Pospisilik JA (2018) The Polycomb-Dependent Epigenome Controls β Cell Dysfunction, Dedifferentiation, and Diabetes. Cell Metabolism 27(6): 1294-1308
- Herman JS, Sagar, Grün D (2018) FateID infers cell fate bias in multipotent progenitors from single-cell RNA-seq data. Nature Methods 15(5): 379-386
- Sagar, Herman JS, Pospisilik JA, Grün D (2018) High-Throughput Single-Cell RNA-Sequencing and Data Analysis. Methods in Molecular Biology 1766: 257-283
- Felmlee D, Grün D, Baumert T (2017) Zooming in on liver zonation. Hepatology 67(2): 784-787
- Tay TL, Mai D, Dautzenberg J, Fernández-Klett F, Lin G, Sagar, Datta M, Drougard A, Stempfl T, Ardura-Fabregat A, Staszewski O, Margineanu A, Sporbert A, Steinmetz LM, Pospisilik JA, Jung S, Priller J, Grün D, Ronneberger O, Prinz M. (2017) A new fate mapping system reveals context-dependent random or clonal expansion of microglia. Nature Neuroscience 20(6): 793-803
- Herrtwich L, Nanda I, Evangelou K, Nikolova T, Horn V, Sagar, Erny D, Stefanowski J, Rogell R, Klein C, Gharun K, Follo M, Seidl M, Kremer B, Münke N, Senges J, Fliegauf M, Aschman T, Pfeifer D, Sarrazin S, Sieweke MH, Wagner D, Dierks C, Haaf T, Ness T, Zaiss MM, Voll RE, Deshmukh SD, Prinz M, Goldmann T, Hölscher C, Hauser AE, Lopez-Contreras AJ, Grün D, Gorgoulis V, Diefenbach A, Henneke P, Triantafyllopoulou, A (2016) DNA Damage Signaling Instructs Polyploid Macrophage Fate in Granulomas. Cell 167(5): 1264-1280
- Muraro MJ, Dharmadhikari G, Grün D, Groen N, Dielen T, Jansen E, van Gurp L, Engelse MA, Carlotti F, de Koning EJ, van Oudenaarden A (2016) A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Systems 3(4): 385-394
- Grün D#, Muraro MJ, Boisset JC, Wiebrands K, Lyubimova A, Dharmadhikari G, van den Born M, van Es J, Jansen E, Clevers H, de Koning EJP, van Oudenaarden A# (2016) De Novo Prediction of Stem Cell Identity Using Single-Cell Transcriptome Data. Cell Stem Cell 19(2): 266-77
- Wu CC, Kruse F, Vasudevarao MD, Junker JP, Zebrowski DC, Fischer K, Noël ES, Grün D, Berezikov E, Engel FB, van Oudenaarden A, Weidinger G, Bakkers J (2016) Spatially Resolved Genome-wide Transcriptional Profiling Identifies BMP Signaling as Essential Regulator of Zebrafish Cardiomyocyte Regeneration. Developmental Cell 36(1): 36-49
- Grün D and van Oudenaarden A. (2015) Design and analysis of single cell sequencing experiments. Cell 163(4): 799-810
- Grün D*, Lyubimova A*, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. (2015) Single-cell mRNA sequencing reveals rare intestinal cell types. Nature 525: 251-255
- Grün D*, Kester L*, van Oudenaarden A. (2014) Validation of noise models for single-cell transcriptomics enables genome-wide quantification of stochastic gene expression. Nature Methods 11(6): 637-40
- Stoeckius M*, Grün D*, Kirchner M, Ayoub S, Torti F, Piano F, Herzog M, Selbach M, Rajewsky N. (2014) Global characterization of the oocyte‐to‐embryo transition in Caenorhabditis elegans uncovers a novel mRNA clearance mechanism. EMBO Journal 33(16): 1751-66
- Stoeckius M*, Grün D*, Rajewsky N. (2014) Paternal RNA contributions in the C. elegans zygote. EMBO Journal 33(16): 1740-50
- Grün D*, Kirchner M*, Thierfelder N*, Stoeckius M, Selbach M, Rajewsky N. (2014) Conservation of mRNA and protein expression during development of C. elegans. Cell Reports 6(3): 565-77
- Kim DH, Grün D, van Oudenaarden A. (2013) Dampening of expression oscillations by synchronous regulation of a microRNA and its target. Nature Genetics 45(11): 1337-44
- Onal P*, Grün D*, Adamidi C*, Rybak A, Solana J, Wang Y, Rahn HP, Chen W, Ziebold U, Rajewsky N (2012) Molecular determinants of pluripotency are deeply conserved between mammalian and planarian stem cells. EMBO Journal 31(12): 2755-69
- Jungkamp AC, Stoeckius M, Mecenas D, Grün D, Mastrobuoni G, Kempa S, Rajewsky N (2011) In vivo and transcriptome-wide identification of RNA-binding protein target sites. Molecular Cell 44 (5): 828-40
- Adamidi C*, Wang Y*, Grün D*, Mastrobuoni G*, You X*, Tolle D, Dodt M, Mackowiak SD, Gogol-Doering A, Oenal P, Rybak A, Ross E, Alvarado AS, Kempa S, Dieterich C, Rajewsky N, Chen W (2011) De novo assembly and validation of planaria transcriptome by massive parallel sequencing and shotgun proteomics. Genome Research 21 (7): 1193-1200
- Grün D, Rajewsky N (2008) Computational prediction of microRNA targets in vertebrates, fruitflies and nematodes. MicroRNAs: From Basic Science to Disease Biology: 172-186
- Lall S*, Grün D*, Krek A, Chen K, Wang YL, Dewey CN, Sood P, Colombo T, Bray N, Macmenamin P, Kao HL, Gunsalus KC, Pachter L, Piano F, Rajewsky N (2006) A genome-wide map of conserved microRNA targets in C. elegans. Current Biology 16 (5): 460-471
- Grün D, Wang YL, Langenberger D, Gunsalus KC, Rajewsky N (2005) microRNA target predictions across seven Drosophila species and comparison to mammalian targets. PLoS Computational Biology 1 (1): e13
- Krek A*, Grün D*, Poy MN*, Wolf R, Rosenberg L, Epstein EJ, MacMenamin P, da Piedade I, Gunsalus KC, Stoffel M, Rajewsky N (2005) Combinatorial microRNA target predictions. Nature Genetics 37 (5): 495-500
Algorithm for rare cell type identification and differentiation trajectory inference from single-cell RNA-seq data
Rosales-Alvarez et al., Genome Biology, 2020; Grün, Nature Methods, 2020; Herman et al., Nature Methods, 2018; Grün et al., Cell Stem Cell, 2016; Grün et al., Nature 2015
Algorithm for scalable inference of lineage-specific gene regulatory networks from single-cell RNA-seq data
Wang et al., Genome Biology, 2024
Algorithm for the inference of cell fate bias in multipotent progenitors from singe-cell RNA-seq data
Herman et al., Nature Methods, 2018
Algorithm for integrative analysis of single-cell RNA-seq and high-resolution spatial transcriptomics data to infer cell state covariation in local niches
Agrawal et al., Nature Communications, 2024
Web interface hematopoietic progenitors
Web interface for the visualization of single-cell RNA-seq data of murine hematopoietic progenitors
Herman et al., Nature Methods, 2018
Web interface for the Human Liver Cell Atlas
Aizarani et al., Nature, 2019
Web interface for the innate lymphocytes from mouse lung and bone marrow
Zeis et al., Immunity, 2020
Web interface for the mouse myocardial infarction atlas
Chan et al., Nature Cardiovascular Research, 2025
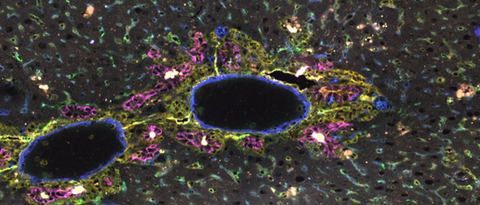
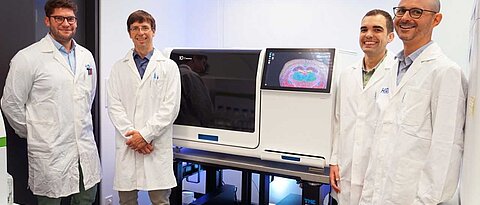

We investigate how signals from the microenvironment are integrated with stochastic processes in a cell to control cell fate decision in normal tissues and upon perturbations due to disease or tissue damage by combining single-cell resolution experimental methods with computational methods involving machine learning and mathematical modeling.
 ERC Consolidator Grant on “Identifying spatial determinants of immune cell fate commitment (ImmuNiche)”")



Prof. Dr. Dominic Grün





Malgorzata Gosia Golda