Current research topics
Our entire research is funded by the DFG
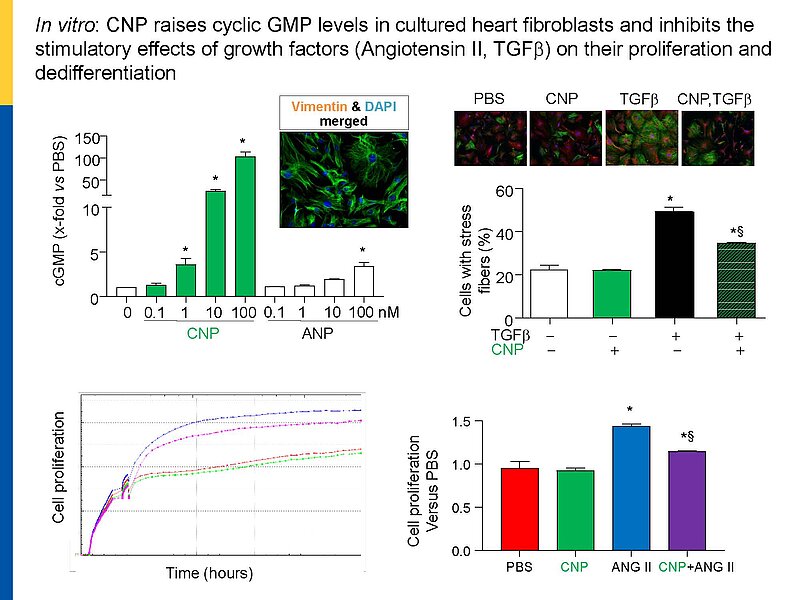
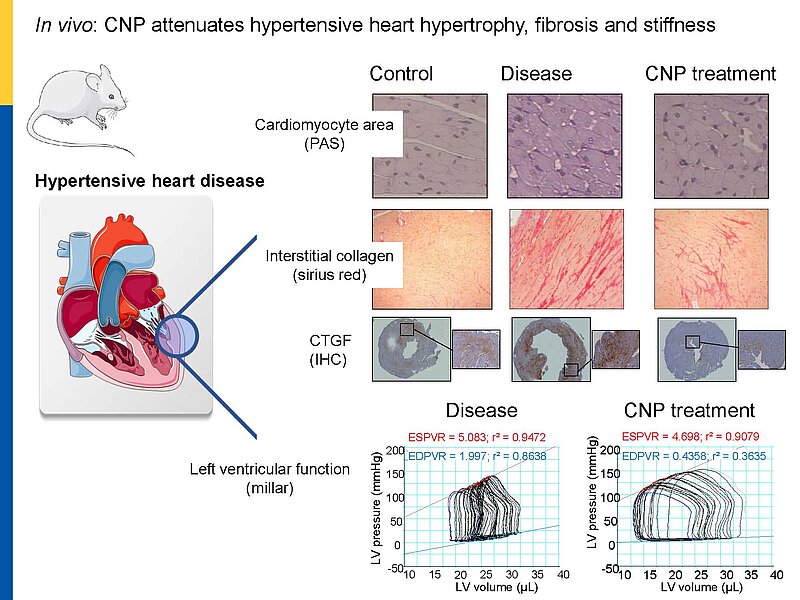
Cardiac remodeling in response to pressure overload or ischemia involves not only cardiomyocyte alterations but also activation of fibroblasts to myofibroblasts and excessive collagen accumulation contributing to impaired cardiac function. These changes are modulated by mechanical forces and neurohumoral factors. Among these factors are the natriuretic peptides atrial and B-type (ANP and BNP, mainly released from myocytes) as well as C-type NP (CNP, secreted by endothelial and infiltrating inflammatory cells). NPs signal through specific transmembrane guanylyl cyclase (GC) receptors and cyclic GMP as second messenger: GC-A (the receptor shared by ANP and BNP) and GC-B (for CNP). Our previous experimental studies in genetic mouse models showed that ANP and BNP may act as key local antihypertrophic and proangiogenic factors during cardiac remodeling. The cardiac role of CNP is less well understood. Its GC-B receptor is expressed in all "resident" myocardial cells (fibroblasts >> cardiomyocytes > vascular cells). Administration of synthetic CNP greatly attenuates cardiac hypertrophy, fibrosis and contractile dysfunction in mice with heart disease.
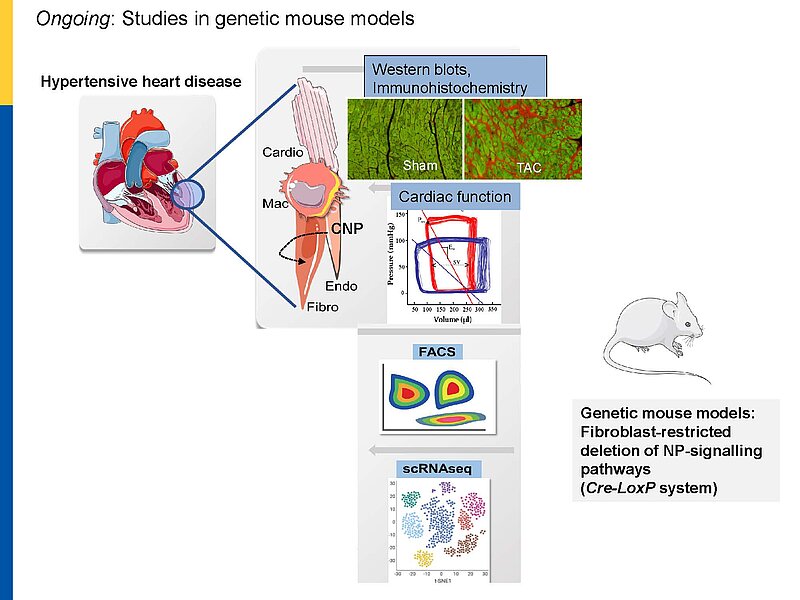
Notably, our previous studies of a novel genetic mouse model with cardiomyocyte-restricted deletion of GC-B indicate that myocytes are only partly involved in these protective CNP actions. In vitro, synthetic NPs (CNP>>ANP) counterregulate the stimulatory effects of TGF-b or Angiotensin II on n fibroblast proliferation, differentiation and collagen secretion.
To dissect the fibroblast actions of endogenous local NPs in vivo, we generated new genetic models with induced, fibroblast-specific deletion of either GC-A or GC-B in adult mice for studies of the impact on hypertensive or ischemic heart disease. Our experimental observations reveal that CNP has local cardioprotective, especially antifibrotic actions.
This may foster clinical studies of CNP or inhibitors of neprilysin-mediated CNP degradation (Sacubitril) in patients with heart diseases such as heart failure with preserved ejection fraction.

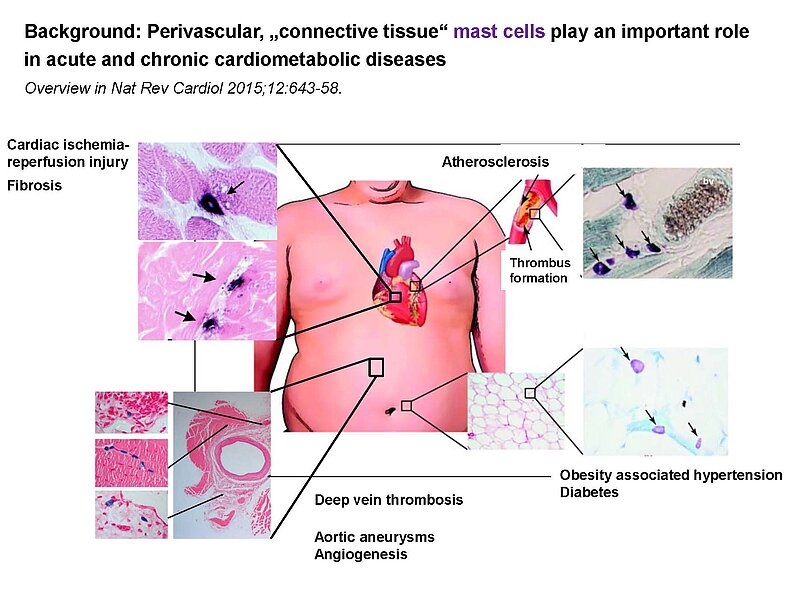
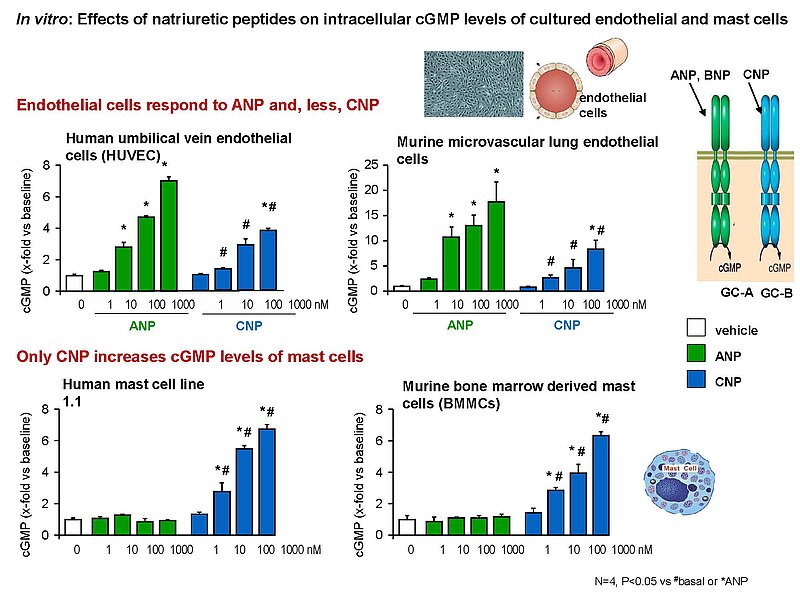
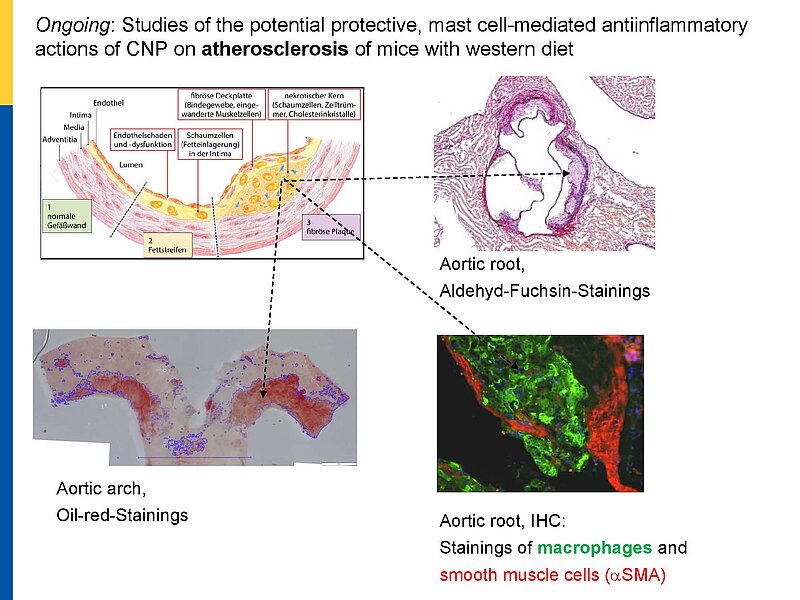
The paracrine communication of endothelial cells (EC) with adjacent luminal, mural and extravascular cells is critically involved in cardiovascular homeostasis. EC dysfunction and altered crosstalk with inflammatory and smooth muscle cells, fibroblasts and myocytes contributes to vascular and cardiac diseases such as atherosclerosis and hypertensive or ischemic heart remodeling. Among these paracrine factors, endothelium-derived C-type natriuretic peptide (CNP) complements the homeostatic actions of nitric oxide and prostacyclin. CNP moderates vascular resistance and thereby systemic blood pressure, and prevents microcirculatory inflammation, atherosclerosis and aneurysms. The cGMP-producing guanylyl cyclase B (GC-B) receptor for CNP is expressed in different vascular and adjacent cells. However, the exact target cells and mechanisms mediating such protective CNP actions are unknown.
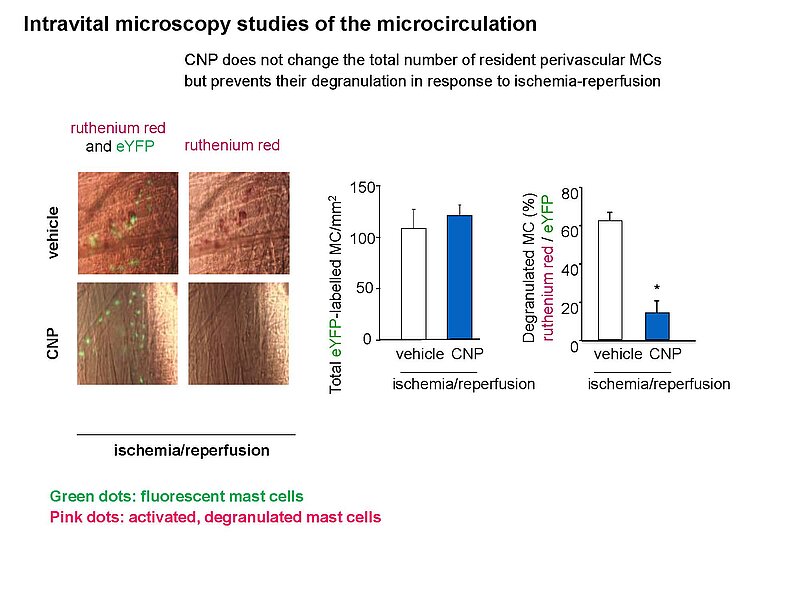
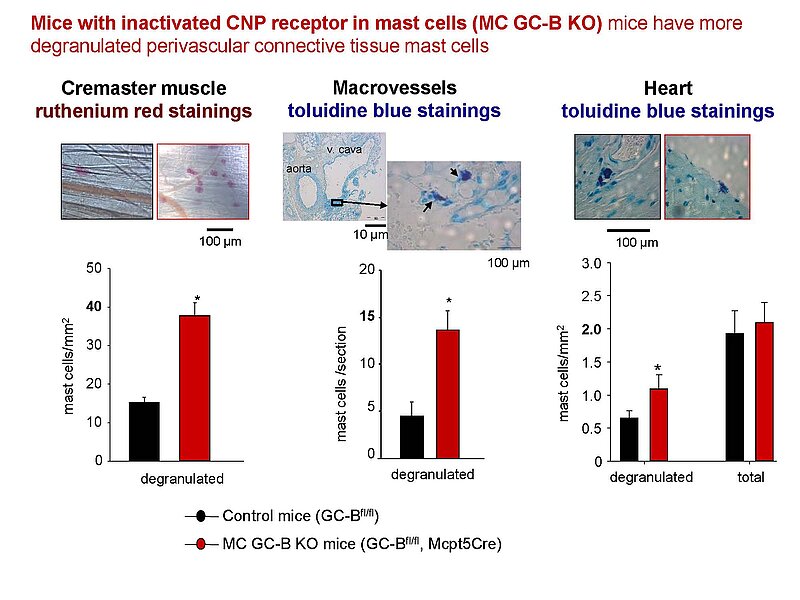
Notably, our ongoing in vitro/in vivo studies reveal CNP-induced GC-B/cGMP signaling in perivascular resident mast cells (MCs). Intravital microscopy studies of the mouse cremaster microcirculation demonstrate that exogenous, synthetic CNP fully prevents ischemia/reperfusion-induced MC degranulation and inflammation. To study whether endogenous, endothelial CNP acts as paracrine modulator of perivascular MCs we generated a novel genetic mouse model with conditional, MC-restricted deletion of the GC-B receptor (MC GC-B KO mice).
In this project we combine studies in MC deficient, MC reporter and MC GC-B KO mice with studies in cultured MCs. Our hypothesis is that endogenous, endothelial CNP counter-regulates the pathologic hyperactivation of (peri)vascular MCs and thereby moderates the development of vascular (atherosclerosis) and/or cardiac inflammation and remodeling (in ischemic or hypertensive heart disease).

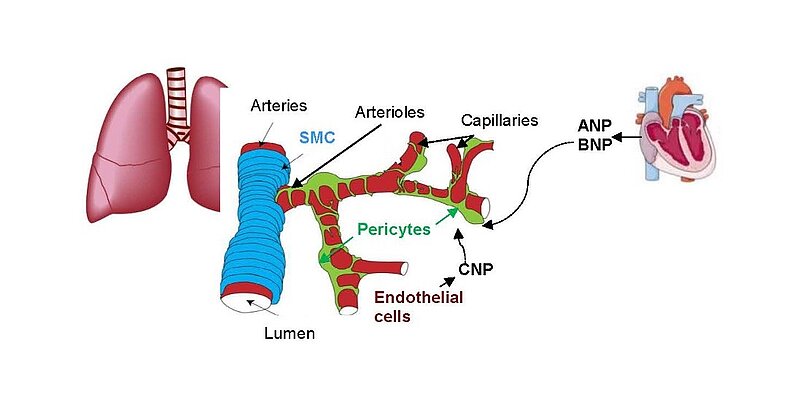
Pulmonary hypertension (PH) affects up to 100 million people worldwide. Despite therapeutical progresses, PH remains a fatal disease with a 3-year survival rate of 58%. Novel therapies are urgently needed.
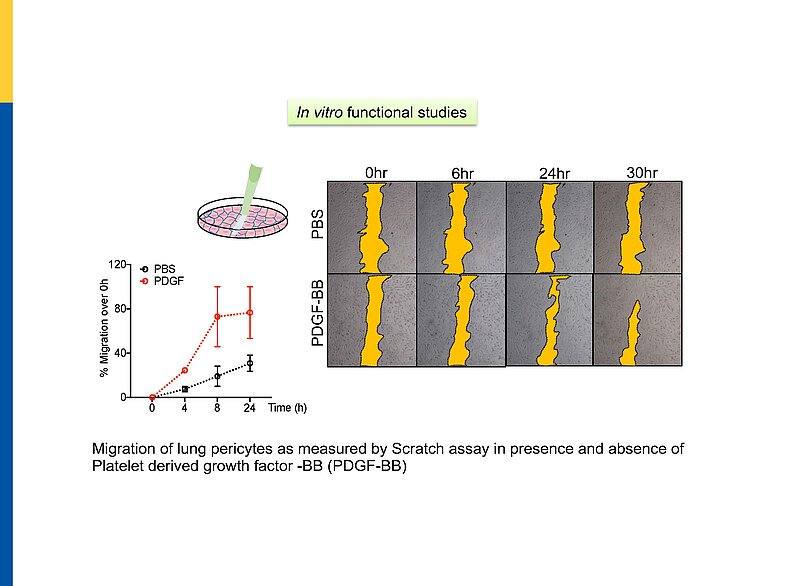
Hypercontraction and loss of pericytes but also their migration, ectopic proliferation and differentiation contribute to microvessel constriction and rarefaction, vascular remodelling and inflammation in PH. While several pathways promoting pericyte dysfunction were described, endogenous factors counter-regulating such changes are barely known. In the systemic and retinal microcirculation ANP- or CNP-induced cGMP signalling reversed ET-1-induced pericyte contractions and thereby arteriolar and capillary constrictions; moreover NPs attenuated hypoxia- and TGF-b-induced pericyte apoptosis, vessel loss and perivascular inflammation Our observation that lung pericytes also express the GC-A and GC-B receptors suggests that pericyte actions of NPs may counterregulate the development of PH. In such case, augmentation of NP/cGMP signalling could be a target for novel therapies of PH.
In this project we address the following hypotheses:
1. Endocrine acting cardiac ANP/BNP and/or paracrine endothelial CNP moderate the contraction of pericytes and diminish pulmonary distal arteriolar and capillary tone. Thereby pericyte-mediated actions of NPs contribute to the maintenance of physiological low PVR. In addition, NPs inhibit the contraction of lung pericytes in response to stressors (hypoxia, ET-1) and pathological increases of PVR.
2. In chronic lung disease (with hypoxia, augmented local levels of vasoconstrictory growth hormones and cytokines) NPs improve lung pericyte´s viability and moderate their proliferation, metabolic changes, dedifferentiation to myofibroblasts and SM-like cells, attenuating PH and right heart disease.
3. At long term, such pathological conditions impair the expression and responsiveness of NP receptors (GC-A, maybe also GC-B) and thereby the protective actions of the endogenous hormones. Inhibition of GC-A or GC-B signalling in pericytes promotes the muscularization of very distal, normally nonmuscularized vessels and perivascular inflammation, thereby contributing to enhanced PVR and PH.
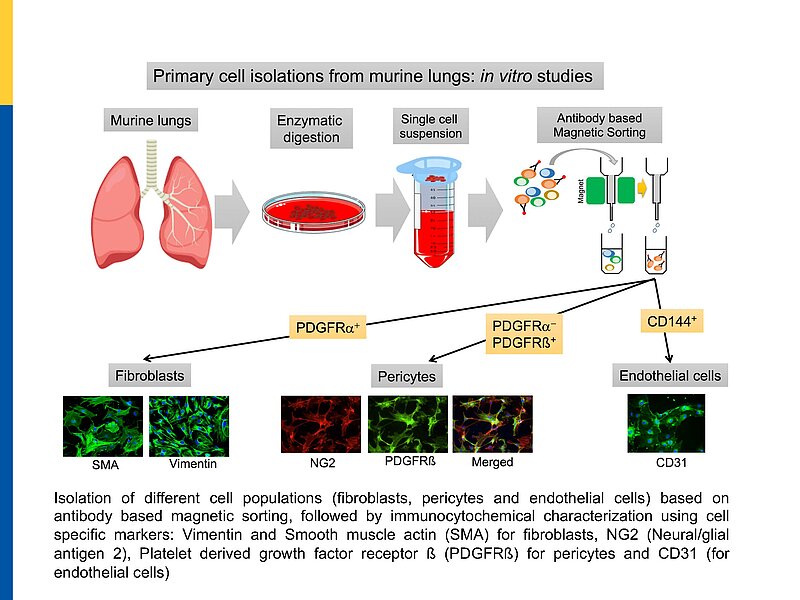
Global GC-A KO or GC-B KO mice have multiple systemic alterations. To bypass these limitations, we generated conditional, pericyte-specific GC-A and GC-B KO lines. To follow the above hypotheses and their relevance for PH we perform comparative studies of pericyte GC-A KO mice, pericyte GC-B KO mice and control littermates. These studies are complemented with in vitro investigations of GC-A/GC-B expression, activity and downstream signalling pathways, i.e. metabolomics in cultured human and murine lung pericytes under physiological and disease conditions.
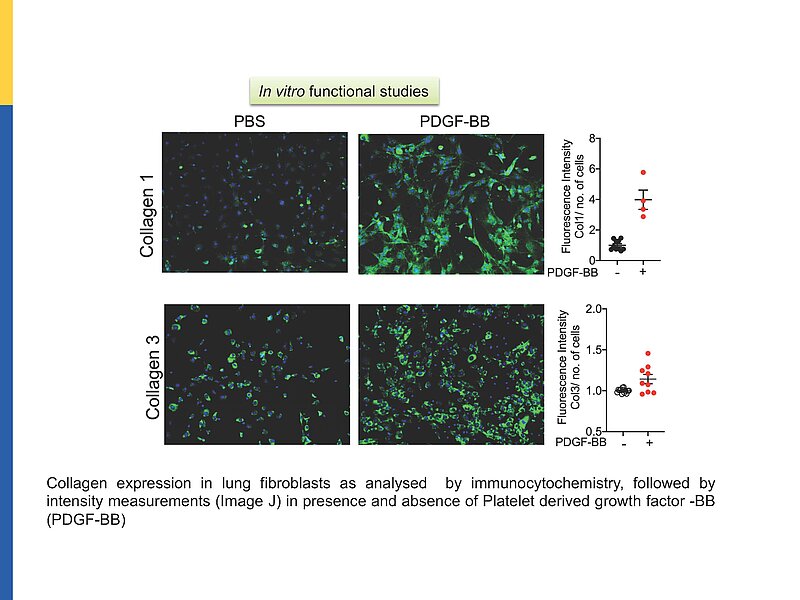
Pulmonary fibrosis (PF) is a lethal, progressive parenchymal lung disorder affecting millions of patients worldwide, with limited therapeutic options. Inflammatory infiltration and fibroblast-to-myofibroblast activation leading to excessive extracellular matrix deposition underlie the pathogenesis of PF.
While several pathways promoting these cellular alterations have been described, endogenous factors counter-regulating such changes are barely known.
Our recent observations in cell culture models suggest that the endothelial hormone C-type natriuretic peptide (CNP) may moderate pathological fibroblast-to-myofibroblast differentiation and thereby lung inflammatory infiltration and fibrosis.
The here proposed studies aim to unravel the mechanisms and clinical implications of this paracrine pathway by employing a novel genetic mouse model and cultured murine and human fibroblasts.
Here you can find the current publications by Dr. Swati Dabral.
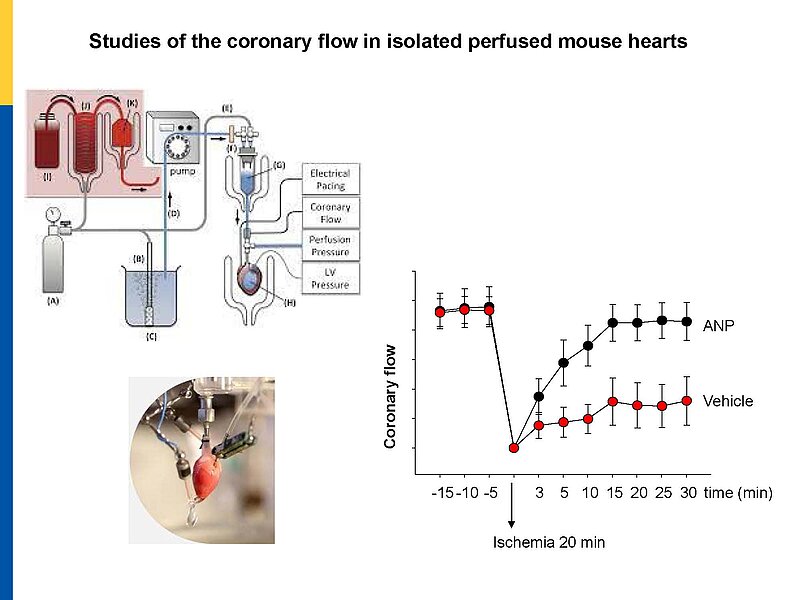
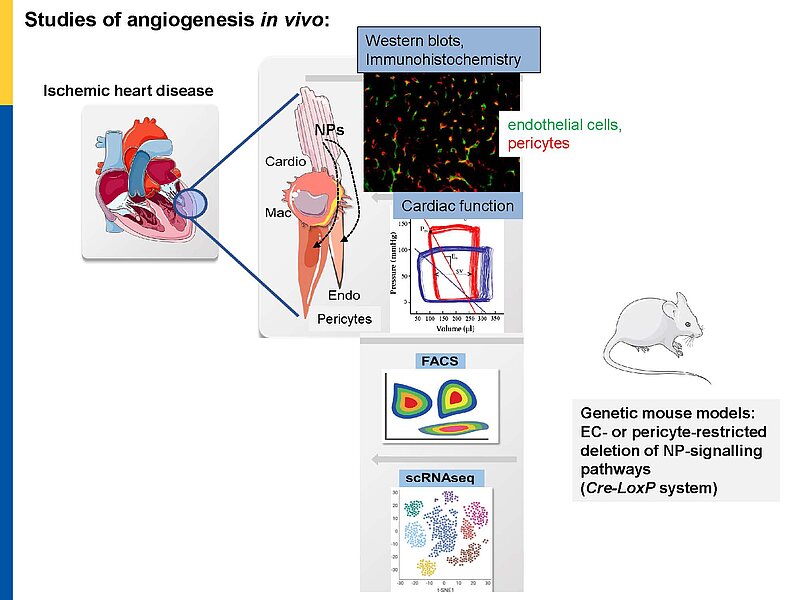
Microvascular dysfunction after acute myocardial infarction (MI) is a major clinical problem. Although primary percutaneous coronary intervention (PCI) has markedly improved patients survival, despite epicardial reperfusion more than 30% of patients show signs of microvascular dysfunction leading to adverse left ventricular remodeling and heart failure. Acutely, vasoconstriction and thrombotic occlusion can reduce microvascular perfusion within the distal myocardium (“no-reflow”). Later, impaired angiogenesis can contribute to myocardial tissue damage. Based on experimental studies, several clinical trials aimed to improve myocardial angiogenesis via intracoronary administration of vascular growth factors, gene transfer or bone marrow mononuclear cells, in patients who had successful primary PCI, but the results were disappointing. A better knowledge of the cellular pathways regulating myocardial (re)perfusion after ischemia is necessary to search for therapeutic strategies capable to restore the microvascular network and flow.
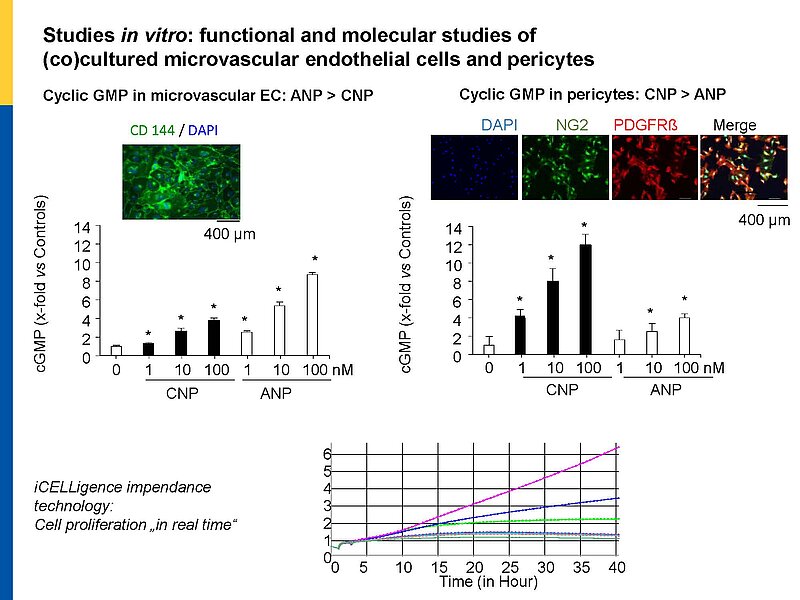
Our project aims to elucidate the role of atrial (ANP) and B-type natriuretic peptides (BNP) in the paracrine communication of cardiomyocytes with coronary microcirculatory endothelial cells and pericytes, as well as the potential pharmacological, therapeutical implications. Based on our previous observations we hypothesize that these cardiac hormones can attenuate acute post-ischemic vasoconstrictions of the coronary microcirculation and improve myocardial angiogenesis.
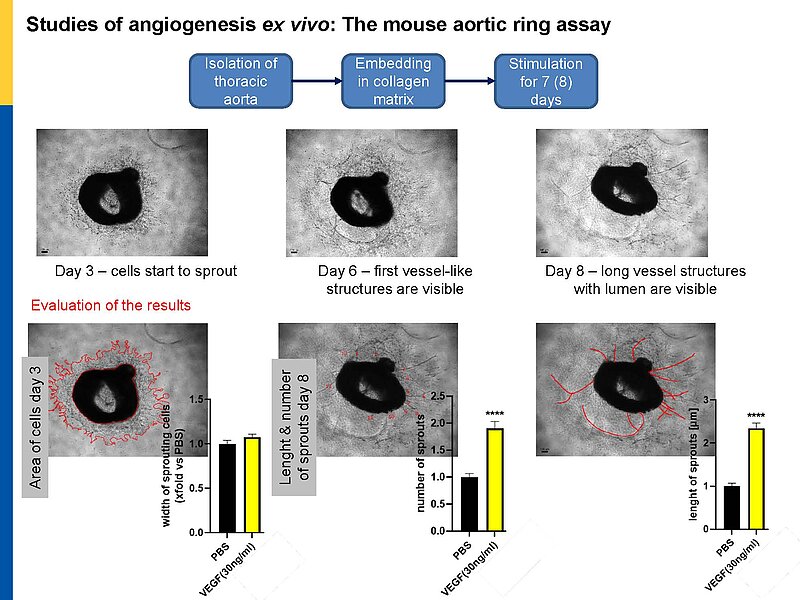
To follow these hypotheses we combine studies of the coronary flow in isolated perfused murine hearts and studies of angiogenesis in vitro, ex vivo and in vivo.