Current research
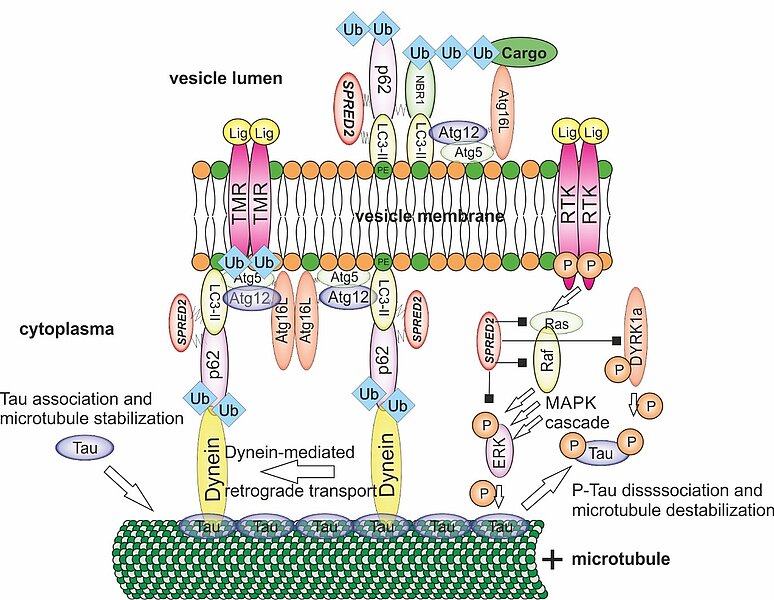
Cardiac functionality is dependent on a balanced protein turnover. Accordingly, regulated protein decay is critical to maintain cardiac function. In this project, we investigate if deficiency of SPRED proteins, an intracellular repressor of ERK-MAPK signaling markedly expressed in human heart, results in impaired autophagy, heart failure, and shortened lifespan. SPRED2-deficiency leads to cardiac hypertrophy and fibrosis and severe arrhythmias. Mechanistically, cardiomyocyte dysfunction results from ERK hyperactivation and dysregulated autophagy, observed as accumulation of vesicles, vacuolar structures, and degenerated mitochondria.
However, the molecular mechanisms how SPRED proteins regulate autophagy remain enigmatic. SPRED proteins have no intrinsic enzymatic activity, which makes an alternative mechanism of regulation of autophagy necessary. We investigate in detail the pathways necessary to clarify how SPRED2 regulates vesicle transport required for signaling but also final signal inactivation by autophagy in cardiac myocytes. Elucidation of these molecular processes in vivo and ex vivo will be relevant in many areas of medical research, as a variety of diseases are triggered by disorders of MAPK pathway inactivation (“Rasopahties”), of intracellular transport, and lack of fusion of lysosomes (“Lysosomal Storage Diseases”).
This project is supported by the DFG.