NGS data analysis
We offer Bioinformatics analysis solutions for high-throughput sequencing, such as RNA-Seq, Dual/Multi RNA-Seq, ChIP-Seq and single cell experiments, including scRNA-seq and ATAC. Our standard analysis includes quality assessment, filtering and trimming of the data.
A properly filled out order sheet or single cell submission sheet is required, in the case of single cell experiments, information on the CMOs, ADTs, and HTOs used is essential.
RIP-seq Example Analysis
Campa AR, Smith LM, Hampton HG, et al. The Rsm (Csr) post-transcriptional regulatory pathway coordinately controls multiple CRISPR–Cas immune systems. Nucleic Acids Research. 2021;49(16):9508-25. https://doi.org/10.1093/nar/gkab704
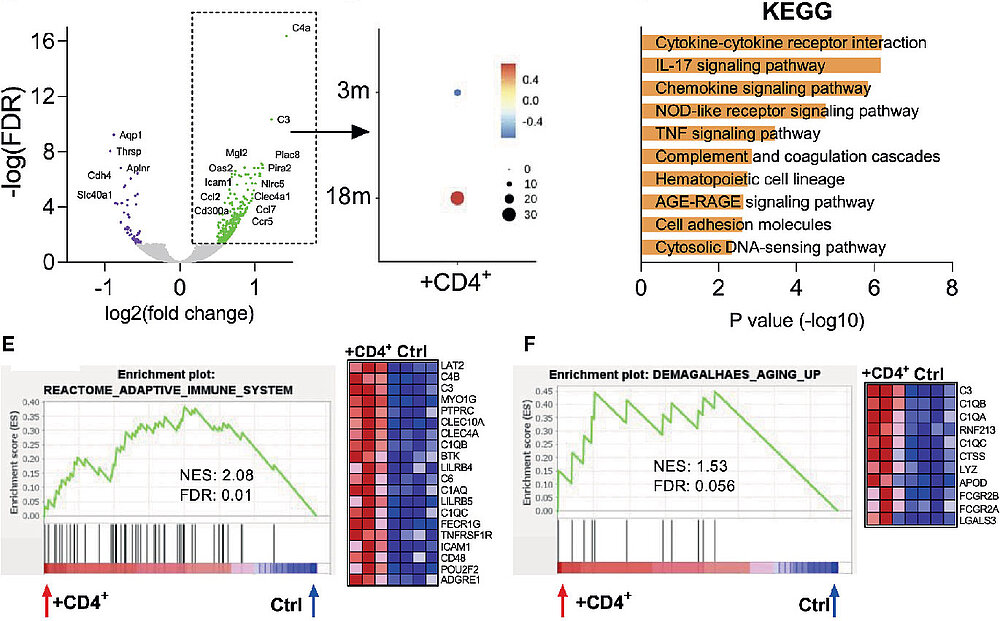
RNA-seq Transcriptome Analysis Example
Delgobo M, Heinrichs M, Hapke N, et al. Terminally Differentiated CD4+ T Cells Promote Myocardial Inflammaging. Front Immunol. 2021;12. https://doi.org/10.3389/fimmu.2021.584538
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality check, Quality and adapter trimming, Alignment, Normalization, Differential gene expression analysis, Functional enrichment analysis |
Raw BCL data, Raw FASTQ files, QC reports, BAM files (opt), Coverage files (opt), Count tables, PCA and sample distance plots, Pairwise comparison tables and plots, Functional enrichment tables and plots |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality check, Quality and adapter trimming, Filtering of host and pathogen reads, Alignment, Normalization, Differential gene expression analysis, Functional enrichment analysis |
Raw BCL data, Raw FASTQ files, QC reports, FastQScreen read tagging reports (opt), BAM files (opt), Coverage files (opt), Count tables, PCA and sample distance plots, Pairwise comparison tables and plots, Functional enrichment tables and plots |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality Check, Trimming, Alignment, Normalization, Peak calling |
Raw BCL data, Raw FASTQ data, BAM files (opt), QC report |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality check, Quality and adapter trimming, De novo genome assembly, Draft genome ordering against a reference (opt), Taxonomic identification (opt), Genome annotation (opt), MLST (opt), Antibiotic resistance genes screening, Virulence genes screening |
Raw BCL data Raw FASTQ files, QC reports, Interactive tormes report, assembly, annotation, taxanomic identification report (opt), MLST, antibacterial and virulence genes (opt) |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality check, Quality and adapter trimming, Computational deconvulation of human and mouse reads, Alignment, Normalization, Differential gene expression analysis, Functional enrichment analysis |
Raw BCL data, Raw FASTQ files, QC reports, XenofilteR report (opt), BAM files (opt), Coverage files (opt), Count tables, PCA and sample distance plots, Pairwise comparison tables and plots, Functional enrichment tables and plots |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality Check, Alignment, Filtering, Barcode counting, UMI counting, Gene Expression |
Raw BCL data, Raw FASTQ data, QC report, Summary reports, BAM, bai files Raw count matrix, Filtered count matrix |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality Check, Alignment, Sequence assembly (V(D)J), Filtering, Barcode counting, UMI counting, Gene Expression Clonotype Calling (V(D)J) |
Raw BCL data, Raw FASTQ data, QC report, Summary reports, BAM, bai files Sample count matrix fasta, fai, annotation, bam and bai files (V(D)J consensus, concatenated V(D)J reference segments), Annotated contigs and consensus sequences of V(D)J rearrangements fasta, fastq and annotation for contigs |
| Analysis steps | Deliverables | ||
|
Demultiplexing, Quality Check, Alignment, Filtering, Barcode counting, UMI counting, Gene Expression |
Raw BCL data, Raw FASTQ data, QC report, Summary reports, BAM, bai files, Peak bed file and annotation, Raw peak bc matrix, Filtered peak bc matrix, Filtered tf bc matrix, Single cell calling |